前言
这一部分内容涉及R中使用人类基因且,内含子,外显子,转录本,AnnotationHub,基因组的注释包,GO分析,KEGG分析等,笔记末尾的参考文献是原文。
基础注释资源与发现
在这一部分里,我们将回顾Bioconductor中用于处理和注释基因组序列的一些工具。我们将研究参考基因组序列,转录本和基因,并以基因通路(gene pathway)作为结束。我们学习这一部分的最终目标就是使用注释信息来帮助我们对基因组实验进行可靠的解释。Bioconductor的基本目标就是更加方便地有关基因组结构和功能的信息统计统计分析程序。
注释概念的层次结构
Bioconductor包括许多不同类型的基因组注释。我们可以在层次结构中来理解这些注释资源。
- 最基因的注释就是某个物种的参考基因组序列。它总是按照核苷酸的线性方式排列成染色体(例如参考基因组。
- 在此之上就是将染色体序列排列到感兴趣的区域中。最感兴趣的区域就是基因,但是注释中也含有其它的信息,例如SNP或CpG位点。基因具有内部结构,即被转录的部分和未被转录的部分。“基因模式”定义了在基因组坐标中的标记和布置这些结构的方式。
- 在感兴趣的区域(regions of interest)的理念下,我们还定义了面向平台的注释(platform-oriented annotation)。这处类型的注释通常首先是由厂家提供的,但随着研究的进行,对这些平台中最初有歧义信息进行了确认和更新,从而完善了这些注释内容。密歇根大学的brainarray project 说明了affymetrix阵列注释的过程。我们将在本节最后讨论面向平台注释的问题。
- 在此之是是将区域(通常是基因或基因的产物)组成成具有共同结构或功能特性的组。例如在细胞中共同被发现的,或者是被鉴定为在生物学过程中协同作用的基因组(我的理解就是GO分析,KEGG分析这一类)。
发现可用的参考基因组
Bioconductor已经包含了注释包的合成,将它这一层次结构上的所有元素都带了可编程环境中。参考基因组序列是使用Biostrings和BSgenome包中的工具进行管理的,available.genomes 函数能够列出构建好的人和现在各种模式生物的参考基因组,如下所示:
|
|
参考基因组的版本很重要
不同物种的参考基因组是从头构建的,然后随着算法和测序数据的不断改进而进一步完善。对人类而言,基因组研究联盟(Genome Research Consortium)于2009年构建了37号版本,并于2013年构建了38号版本。
一旦参考基因组构建完成,就哦可以很轻松地对某个物种进行信息丰富的基因组序列分析,因为人们可以专注于那引起已知含有等位基因多样性的区域。
The reference build for an organism is created de novo and then refined as algorithms and sequenced data improve. For humans, the Genome Research Consortium signed off on build 37 in 2009, and on build 38 in 2013.
需要注意的是,基因组序列包含有很长的名称,这个名称里包括版本信息。这样命名的方式就是为了避免与不同版本的参考基因组混淆。在LiftOver这节视频里,我们就展示了如何使用UCSC的liftOver工具与rtracklayer包中的接口对接,从而实现不同版本的基因组坐标转化的过程。
为了帮助用户避免混淆从不同参考基因组坐标上收集分析来的数据,我们提供了一个”基因组“标签,这个标签填充了大多关于序列的信息。在随后的部分里,我们会看到一些案例。用于序列比对的软件可以检查被比对上的序列的兼容标签,从而有助于确保有意义的结果。
H. sapiens的参考基因序列
通过安装并添加一个单独的R包就能获取智人(Homo sapiens)的参考序列。这个程序包定义了一个Hsapiens对象,试剂公司对象是染色体序列的来源,但是当对其进行单独显示时,它会提供相关序列数据来源的信息,如下所示:
|
|
我们使用 $ 符号来获取17号染色体的序列,如下所示:
|
|
参考序列的转录本和基因
UCSC注释
TxDb包家族和数据对象管理了转录本和基因模式信息。我们可以认为这些信息来源于UCSC基因组浏览器的注释表,如下所示:
|
|
我们使用 genes() 来获取Entrez Gene ID的地址,如下所示:
|
|
我们也可以使用合适的标识符进行信息过滤。现在我们提取两个不同基因的外显子,这些外显子由其Entrez基因ID标明,如下所示:
|
|
ENSEMBL注释
Ensembl home主页上写道:Ensembl创建,整合和发布研究基因组的参考数据库和工具。该项目位于 欧洲分子生物学实验室,该实验室的数据库支持其注释资源可以与Bioconductor兼容。
ensembldb 包含有一个简要说明,其内容如下所示:
ensembldb包提供了一些函数,这些函数用于创建和使用以转录本为中心的注释数据库/包。使用注释数据库的Perl API可以从Ensembl 1中直接获取这些数据。TxDb 包的功能和数据类似于GenomicFeatures包,另外,除了从数据库检索所有的基因/转录本模型和注释外,ensembldb包还提供了一个过滤框架,用于检索特定条目的注释,例如位于染色体区域上的某编码基因或某LincRNA转录模式的特定条目。从1.7版本开始,由ensembldb创建的EnsDb数据库还包含蛋白质注释数据库(参考第11节:数据库而已和可用属性/列的概述)。有关蛋白质注释的信息请参考蛋白质的vignette,如下所示:
|
|
举例说明如下:
|
|
你的数据将会成他人的注释:导入/导出
ENCODE项目很地说明了今天的实验是明天的注释。你应该以同样的方式考虑自己的实验(当然,要使实验成为可靠且持久的注释,它必须解决有关基因组结构或功能的重要问题,并且必须使用适当的,能正确执行的实验流程。需要注意,ENCODE能够非常明确地将实验流程链接到数据)。
例如,我们来看一个雌激素受体结合数据,它是由ENCODE发布的一个narrowPeak 数据。它的碱基是用ascii文本表示的,因此可以很容易地导入为一组文本数据。如果记录的字段有一定的规律性,则可以将文件作为表格导入。
但是,我们不仅是想导入数据,还想将导入的数据作为可计算的对象。我们认识到arrowePeak和bedGraph格式之间的联系后,我们就可以立即将其导入GRanges中。
为了说明这一点,我们在ERBS包中找到narrowPeak原始数据文件的路径,如下所示:
|
|
使用import命令非常简单,如下所示:
|
|
我们可以通过一次获取GRanges。元数据列中还有一些其他字段用于指定名称,但是如果我们只对范围感兴趣,除了添加基因组元数据以防止与不兼容的坐标中记录的数据非法组合外,我们就完成了这个任务(这一段不太理解,原文如下):
We obtain a GRanges in one stroke. There are some additional fields in the metadata columns whose names should be specified, but if we are interested only in the ranges, we are done, with the exception of adding the genome metadata to protect against illegitimate combination with data recorded in an incompatible coordinate system.
为了与其他得养家或系统进行交流,我们有两个主要选择。我们可以将GRanges保存为RData对象,轻松地传递给另外一个R用户使用。或者,我们歌词采用其他标准格式进行导出。例如,如果我们仅对间隔地址和绑定的得分感兴趣,则仅保存为“bed”格式就足够了,如下所示:
|
|
我们已经进行了导入,建模和导入实验数据之间的“往返”,该实验数据可以与其他数据集成在一起,从而增进生物学的理解。
我们需要注意的是,注释在某种程度上是永久正确的,它与在知识边界上的研究进展乏味地隔离开来。我们已经看到了,甚至人类染色体的参考序列也受到了修订。在使用ERBS包时,我们将未知的实验结果视为定义ER结合位点从而进入潜在的生物学解释。不确定性,峰鉴定的可变质量,尚未得到明确估计,但应该是这个样子。
Bioconductor已经尽力致力于这种情况的多个方面。我们维护软件先前版本和注释的存档,以便可以检查或修改过去的工作。我们每年会两次更新中疏注释资源,以确保正在进行的工作以及获得新知识的稳定性。而且,我们已经简化了导入和创建实验数据和注释数据的表示形式。
AnnotationHub
AnnotationHub包用于获取GRanges或其它的合适设计的容器,用于机构设计的容器,如下所示:
|
|
我们可以通过AnnotationHub获得许多与HepG2细胞系相关的实验数据对象,如下所示:
|
|
query 方法可以使用过滤字符串的向量。要限制对寻址组蛋白H4K5的注释资源的响应,只需要添加该标签,如下所示(To limit response to annotation resources addressing the histone H4K5, simply add that tag):
|
|
The OrgDb基因注释图
那些命名为org.*.ge.db 的包含在基因水平上链接到位置,蛋白产物标识符,KEGG途径和GO term,PMIDs以及其它注释资源的标识符的信息,如下所示:
|
|
基因集和通路资源
基因本体论
Gene Ontology (GO)是一种广泛使用的结构化词汇,它组织了基因和基因产物在以下方面的内容:
- 生物过程
- 分子功能
- 细胞组分。
这套词汇本身旨在与所有生物有关。它采用有向无环图的形式,其中term作为节点,使用is-a和part-of关系作构成了大多数链接。
将生物体特定基因链接到基因本体中的术语的注释与词汇表本身是分开的,并且涉及不同类型的证据。这些记录都在Bioconductor的注释包中。
我们可以使用GO.db包来快速地访问GO词汇,如下所示:
|
|
使用AnnotationDbi包中的keys,columns和select函数也很容易在地id与不同terms之间进行映射,如下所示:
|
|
词汇表的图形结构被编码在SQLite数据库的表中。我们可以使用RSQLite接口对此进行查询,如下所示:
|
|
以下查询提示了一些内部标识符:
|
|
我们可以将 mitochondrion inheritance term追溯到父项和祖父母项,如下所示:
|
|
将 “mitochondrion inheritance” 视为过程“mitochondrion distribution”和 “organelle inheritance”在概念上的精练是有意义的,这两个term在数据库中被为父项。
可以使用 GO_dbschema()来查看整个数据库模式。
KEGG
自Bioconductor诞生以来,KEGG的注释就能在Bioconductor中人使用了,但KEGG的数据库使用权限已经进行了更改。当我们使用KEGG.db加载后会出现以下信息,如下所示:
|
|
因此我们可以关注KEGGREST这个包,它需要联网。这是一个非常有用的,基于Entrez标识符的工具。现在我们查询一下BRCA2的信息(它的EntrezID为675),如下所示:
|
|
我们也可以通过keggGet函数来获取构成通路模式的基因列表,如下所示:
|
|
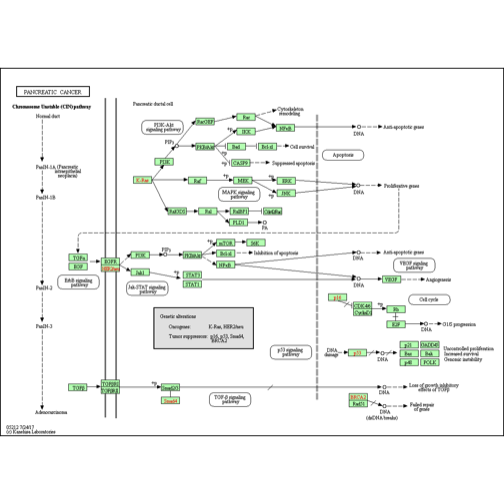
KEGGREST还有许多值得研究的地方,例如还可以查询BRCA2(人类)关于胰腺癌途径的静态图像,如下所示:
|
|
其它本体
rols包含有与EMBL-EBI连接的接口 Ontology Lookup Service.
|
|
为了控制查询检索中涉及的网络流量,搜索分为几个阶段,如下所示:
|
|
ontologyIndex 包支持导入开放生物本体(OBO, Open Biological Ontologies)格式的数据,并含有用于查询和可视化本体系统高效的工具。
通用基因集管理
GSEABase 包有一个用于管理基因集和集合的优秀工具。我们可以从MSigDb中导入胶质母细胞瘤相关的基因集来说明一下,如下所示:
|
|
模式生物的统一,自我描述方法
OrganismDb包简化了对注释的访问。还可以针对TxDb和org.[Nn].eg.db进行直接查询,如下所示:
|
|
面向平台的注释
通过在NCBI GEO的GPL信息页面 上对信息进行排序,我们就可以看到最常用的寡核苷阵列平台(数据库中有4760个系列)就是Affy Human Genome U133 plus 2.0 array (GPL 570)。我们可以使用hgu133plus2.db对这些数据进行注释,如下所示:
|
|
这个资源(以及ChipDb类的所有实例)的基本目的是在探针集(probeset)识别符和更高层次的基因组注释之间进行映射。
有关探针的详细信息(探针集的组成部分)已经由那些后缀为probe的文件包提供,如下所示:
|
|
将探针集标识符映射到基因水平的信息可以提示一些有意思的歧视,如下所示:
|
|
显然,该探针集合可以用于mRNA和miRNA丰度的定量。作为稳定的检查,我们可以看到,不同的符号映射到了相同的细胞带(最后一句不懂,原文为: As a sanity check we see that the distinct symbols map to the same cytoband)。
总结
我们现在已经拥有了含有从核酸到通路水平的许多数据。通过Bioconductor.org上的View就可以查看现有的一些资源。